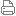
| |






《iPhone11》更新15.1rc。电池效率85%。图里充满100电开始更新15.1rc然后一直用到现在,这续航算鼎的吧 ...





开盘数据恢复成功 设备:WD10JMVM SN:WXC1EC2DH295 故障:硬盘磁头损坏 解决方案:南京西数工程师通过检测及专业设备 ...



电话 15756447447 微信 15756447447 成都诚裕典当,全天营业 成都地区24小时江湖救急,义不容辞! 典当/回收业务范围 ...


手持iPhone 12打《原神》掉帧到20?iPad Pro剪辑4K视频闪退?升级设备动辄花费上万! 2025年实测数据显示:83%的苹果 ...



程绍正韬写字楼作品 顶级豪宅大师『程绍正韬』写字楼首发作品 蔡屋围建面约128-1400㎡home office现正发售 image.png ...

💰出售信和一期三室132.5平85.5万可议精装婚房,品牌家电阳卧阳厅双卫明厨明卫最好楼层,老证一手。电话15003271133+1 ...
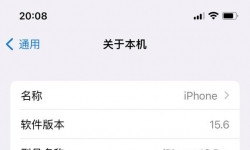

最新版本 macOS Tahoe 26 B3 版本,安装成功。声显网正常。使用正常。 最新系统macOS Tahoe 26 Beta3 官方下载地址: ...






飞利浦推出了新款24寸显示器——24M2N5200X,售价5999元。 新款显示器配备了一块24.1英寸的IFast-TN面板,分辨率为192 ...



微软近日发布 Windows 11、Windows 10 系统更新,在桌面整合了 Edge 搜索栏。微软官方表示引入该功能,其目的是简化用 ...

内存卡是京东麒麟512g,去年朋友带的全新美版机器,已贴钢化膜,电池健康度百分之102,带一个原装Switch pro手柄带tom ...

steam上wows更新花了70G流量,更新下完重下重复三次屁用没有。想试试用epic下,只要绑定的wg账号和区服一样数据应该是 ...





游戏时间站[yxsjz.cn],电脑vr游戏下载,Steamvr游戏,oculus Quest游戏,rifts游戏下载。 中文VR游戏,更有vr一体机 ...
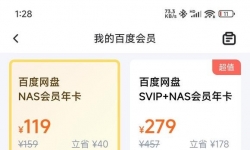
h99pro的79元的百度云nas会员下载百度网盘2.0只有5m速度,到期后现在涨价119元了。不知道是不是我的机器性能太垃圾, ...

惠普 Z4 G5 图形工作站!专业应用的不二之选 💼🎯 北京台式工作站总代理商 | 🔧 北京台式工作站厂家定制首选!在如今 ...




甲苯试听索尼黑砖二代、zx706、飞傲M23 首先感谢甲苯提供的线下试听,买了ie900有两个月了,之前一直搭配飞傲KA17使 ...

FL Studio 24中文版破解版是一款专为音乐创作者和制作人设计的移动端音乐制作软件,它集成了丰富的音频编辑、混音、录 ...

San Diego Comic Con 2025 continues! Thanks to our TFW2005 team at the event, we have new images of Hasbro Tran ...






 微信扫一扫
微信扫一扫